
PRODUCTS and MEDICINES:
Texas Compassionate Use Program (TCUP) - Qualifying Conditions

The Texas medical cannabis program is called the Texas Compassionate Use Program (TCUP). TCUP is administered by the Texas Department of Public Safety (DPS) under the authority of the Texas Health and Safety Code, Chapter 487.
Number 1 question about TCUP: What are the qualifying conditions to become a registered patient in TCUP?
Currently, there is a list of over one hundred conditions that qualify Texas patients to register with TCUP. Diagnosis of one of these conditions must be made by a certified medical practitioner, and then a medical cannabis practitioner (a separate specialist) can determine whether and what to prescribe. The prescription will be one of the products that is legally available through TCUP. Below is a simple graphic on the four steps that a Texas qualifying patient needs to take to become a registered TCUP patient.
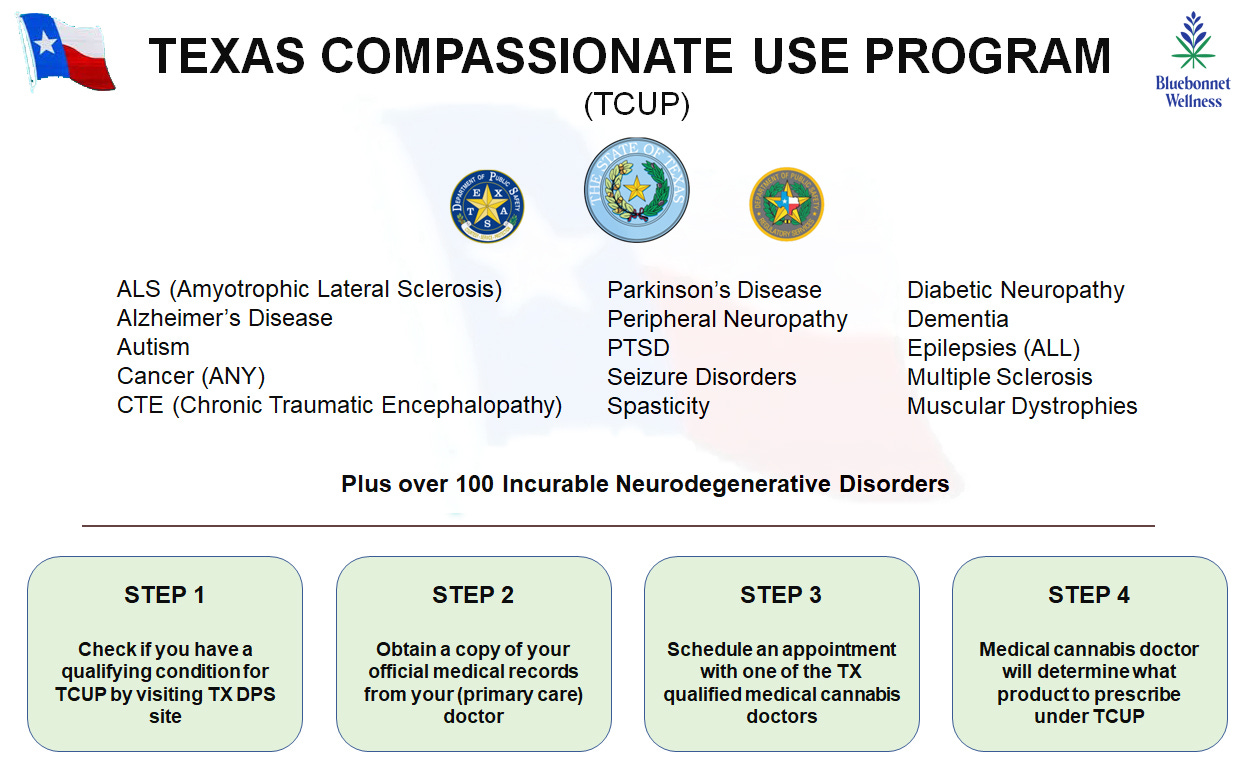
Number 2 question about TCUP: What products are available to registered TCUP patients?
Only ingestible products are currently available through TCUP, which means all the edible products, including gummies. Other popular ingestible products include cannabis oils that are made from cannabis flowers. Ingestible products, like oils, gummies, lozenges, or drinks take time before there is an onset of the effects – medicinal or psychotropic. These types of products enter the digestive system and even small doses of delta-9 THC can cause a significant ‘high’. This happens because there are secondary metabolites of THC that are produced by our own bodies, which also causes the additional ‘high’. In fact, we have a whole article on different ways of consuming cannabis coming out very soon!
Number 3 question: What does 1% THC limit mean?
One percent THC means that the final product cannot contain more than 10 milligrams (mg) of THC per one gram of oil, or edible mass. Recall from above that this limit means that one gummy dose or one milliliter of oil from the oil dropper cannot contain more than 10mg of THC. This is a significant dose for most folks and can get a more sensitive person ‘high’. However, for medicinal purposes to treat severe pain and other symptoms of qualifying disorders, a patient may need to consume tens or even hundreds of milligrams of THC. When cannabis is consumed via the inhalation, then the secondary metabolites that we discussed above do not form. In general, inhalation is a better controlled manner of managing the unwanted side effects of cannabis, such as feeling medicated and too high’ or even paranoid. For now, TCUP does not allow for any inhalation products, but based on the surrounding states and other global programs, this will likely change.
Below is the full list of all of the qualifying TCUP conditions:
ALS (Amyotrophic Lateral Sclerosis)
Alzheimer’s Disease
Autism
Cancer (ANY)
CTE (Chronic Traumatic Encephalopathy)
Diabetic Neuropathy*
Dementia
Epilepsies (ALL)
Multiple Sclerosis
Muscular Dystrophies
Neurodegenerative Diseases
Parkinson’s Disease
Peripheral Neuropathy
PTSD
Seizure Disorders
Spasticity
For more information on TCUP by DPS, click here: TCUP by DPS
Here is the complete list of the qualifying neurodegenerative disorders:
(1) Incurable Neurodegenerative Diseases with Adult Onset:
(A) Motor Neuron Disease:
(i) Amyotrophic lateral sclerosis;
(ii) Spinal-bulbar muscular atrophy; and
(iii) Spinal Muscular Atrophy.
(B) Muscular Dystrophies:
(i) Duchenne Muscular Dystrophy;
(ii) Central Core; and
(iii) Facioscapulohumeral Muscular Dystrophy.
(C) Freidreich's Ataxia.
(D) Vascular dementia.
(E) Charcot Marie Tooth and related hereditary neuropathies.
(F) Spinocerebellar ataxia.
(G) Familial Spastic Paraplegia.
(H) Progressive dystonias DYT genes 1 through 20.
(I) Progressive Choreas: Huntington's Disease.
(J) Amyloidoses:
(i) Alzheimer's Disease;
(ii) Prion Diseases:
(I) Creutzfeldt-Jakob Disease;
(II) Gerstmann-Straussler-Scheinker Disease;
(III) Familial or Sporadic Fatal Insomnia; and
(IV) Kuru.
(K) Tauopathies.
(i) Chronic Traumatic Encephalopathy:
(ii) Pick Disease;
(iii) Globular Glial Tauopathy;
(iv) Corticobasal Degeneration;
(v) Progressive Supranuclear Palsy;
(vi) Argyrophilic Grain Disease;
(vii) Neurofibrillary Tangle dementia, also known as Primary Age-related Tauopathy; and
(viii) Frontotemporal dementia and parkinsonism linked to chromosome 17 caused by mutations in MAPT gene.
(L) Synucleinopathies:
(i) Lewy Body Disorders:
(I) Dementia with Lewy Bodies; and
(II) Parkinson's Disease; and
(ii) Multiple System Atrophy.
(M) Transactive response DNA-binding protein-43 (TDP-43) Proteinopathies:
(i) Frontotemporal Lobar Degeneration;
(ii) Primary Lateral Sclerosis; and
(iii) Progressive Muscular Atrophy.
(2) Incurable Neurodegenerative Diseases with Pediatric Onset:
(A) Mitochondrial Conditions:
(i) Kearn Sayers Syndrome;
(ii) Mitochondrial Encephalopathy Ragged Red Fiber;
(iii) Mitochondrial Encephalopathy Lactic Acidosis Stroke;
(iv) Neuropathy, Ataxia, and Retinitis Pigmentosa;
(v) Mitochondrial neurogastrointestinal encephalopathy;
(vi) Polymerase G Related Disorders:
(I) Alpers-Huttenlocher syndrome;
(II) Childhood Myocerebrohepatopathy spectrum;
(III) Myoclonic epilepsy myopathy sensory ataxia; and
(IV) Ataxia neuropathy spectrum;
(vii) Subacute necrotizing encephalopathy, also known as Leigh syndrome;
(viii) Respiratory chain disorders complex 1 through 4 defects: Co Q biosynthesis defects;
(ix) Thymidine Kinase;
(x) Mitochondrial Depletion syndromes types 1 through 14:
(I) Deoxyguanisine kinase deficiency;
(II) SUCLG1-related mitochondrial DNA depletion syndrome, encephalomyopathic form with methylmalonic aciduria; and
(III) RRM2B-related mitochondrial disease.
(B) Creatine Disorders:
(i) Guanidinoacetate methytransferase deficiency;
(ii) L-Arginine/glycine amidinotransferase deficiency; and
(iii) Creatine Transporter Defect, also known as SLC 6A8.
(C) Neurotransmitter defects:
(i) Segawa Diease, also known as Dopamine Responsive Dystonia;
(ii) Guanosine triphosphate cyclohydrolase deficiency;
(iii) Aromatic L-amino acid decarboxylase deficiency;
(iv) Monoamine oxidase deficiency;
(v) Biopterin Defects:
(I) Pyruvoyl-tetahydropterin synthase;
(II) Sepiapterin reductase;
(III) Dihydropteridine reductase; and
(IV) Pterin-4-carbinolamine dehydratase.
(D) Congenital Disorders of Glycosylation.
(E) Lysosomal Storage Diseases:
(i) Mucopolysaccaridosis:
(I) Mucopolysaccharidosis Type I, also known as Hurler Syndrome or Scheie Syndrome;
(II) Mucopolysaccharidosis Type II, also known as Hunter Syndrome;
(III) Mucopolysaccharidosis Type III, also known as Sanfilippo A and B;
(IV) Mucopolysaccharidosis Type IV, also known as Maroteaux-Lamy; and
(V) Mucopolysaccharidosis Type VII, also known as Sly.
(ii) Oligosaccharidoses:
(I) Mannosidosis;
(II) Alpha-fucosidosis;
(III) Galactosialidosis;
(IV) Asparylglucosaminuria;
(V) Schindler; and
(VI) Sialidosis;
(iii) Mucolipidoses:
(I) Mucolipidoses Type II, also known as Inclusion Cell disease; and
(II) Mucolipidoses Type III, also known as pseudo-Hurler polydystrophy;
(iv) Sphingolipidoses:
(I) Gaucher Type 2 and Type 3;
(II) Neimann Pick Type A and B;
(III) Neimann Pick Type C;
(IV) Krabbe;
(V) GM1 gangliosidosis;
(VI) GM2 gangliosidosis also known as Tay-sachs and Sandhoff Disease;
(VII) Metachromatic leukodystrophy;
(VIII) Neuronal ceroid lipofuscinosis types 1-10 including Batten Disease; and
(IX) Farber Disease; and
(v) Glycogen Storage-Lysosomal: Pompe Disease.
(F) Peroxisomal Disorders:
(i) X-linked adrenoleukodystrophy;
(ii) Peroxisomal biosynthesis defects:
(I) Zellweger syndrome:
(II) Neonatal Adrenoleukodystrophy; and
(iii) D Bidirectional enzyme deficiency.
(G) Leukodystrophy:
(i) Canavan disease;
(ii) Pelizaeus-Merzbacher disease;
(iii) Alexander disease;
(iv) Multiple Sulfatase deficiency;
(v) Polyol disorders;
(vi) Glycine encephalopathy, also known as non-ketotic hyperglycinemia;
(vii) Maple Syrup Urine Disease;
(viii) Homocysteine re-methylation defects;
(ix) Methylenetetrahydrofolate reductase deficiency severe variant;
(x) L-2-hydroxyglutaric aciduria;
(xi) Glutaric acidemia type 1;
(xii) 3-hydroxy-3-methylglutaryl-CoA lyase deficiency;
(xiii) Galactosemia;
(xiv) Manosidosis alpha and beta;
(xv) Salidosis;
(xvi) Peripheral neuropathy types 1 through 4;
(xvii) Pyruvate Dehydrogenase Deficiency;
(xviii) Pyruvate Carboxylase Deficiency;
(xix) Refsum Disease; and
(xx) Cerebral Autosomal Dominant Arteriopathy with Sub-cortical Infarcts and Leukoencephalopathy.
(H) Fatty Acid Oxidation:
(i) Trifunctional protein deficiency; and
(ii) Long-chain L-3 hydroxyacyl-CoA dehydrogenase deficiency.
(I) Metal Metabolism:
(i) Wilson Disease;
(ii) Pantothenate Kinase Associated Neurodegeneration; and
(iii) Neurodegeneration with brain iron accumulation.
(J) Purine and Pyrimidine Defects:
(i) Adenylosuccinate synthase Deficiency;
(ii) 5-aminoimidazole-4-carboxamide ribonucleotide transformylase deficiency;
(iii) Hypoxanthine-guanine phosophoribosyltransferase Deficiency also known as Lesch-Nyhan disease;
(iv) Dihydropyrimidine dehydrogenase Deficiency; and
(v) Dihydropirimidinase Deficiency.
Copyright Bluebonnet Wellness 2021 © by Adendox, LLC and Dr. Jokubas Ziburkus PhD aka Dr. Z